|
AutoDock Vina 1. Overview and InstallationAutoDock Vina [1] is a powerful open-source program designed by Dr. Oleg Trott at The Scripps Research Institute for molecular docking tasks. Official Website: https://vina.scripps.edu/ Download Page: https://vina.scripps.edu/downloads/ AutoDock Vina can be installed on various operating systems including Linux, MAC, and Windows. For Linux users, the installation can also be performed via the terminal using the following command: sudo apt install autodock-vina For additional guidance and tutorials, a video tutorial is available on YouTube: https://www.youtube.com/watch?v=-GVZP0X0Tg8. Setting up the docking calculations requires the installation of supporting software: MGLTools [2]: This tool is essential for the setup and analysis of docking calculations. Download it from the official MGLTools Downloads page, suitable for your operating system. https://ccsb.scripps.edu/mgltools/downloads/ Open Babel [3]: Open Babel is utilized to convert output files from the docking program to the PDB file format. Install Open Babel by following the instructions provided here. https://open-babel.readthedocs.io/en/latest/Installation/install.html For Linux installations, Open Babel can be installed using the following command: sudo apt install openbabel Rasmol for Visualization: Rasmol is employed for visualizing PDB structures. Obtain Rasmol from its official webpage: For Linux installations, execute the following command to install Rasmol: sudo apt install rasmol The combination of AutoDock Vina, MGLTools, Open Babel, and Rasmol provides a comprehensive set of tools necessary for successful molecular docking experiments. 2. Acquiring PDB Structures for DockingTo perform docking simulations of a ligand in a receptor, you'll require PDB structures for both species involved. The ligand structure can be constructed using various programs such as GaussView or Avogadro [4]. It's advisable to optimize the ligand structure using Gaussian or ORCA QM packages for better accuracy. After optimization, reconvert the output file to the PDB format using Open Babel. Obtain the receptor's PDB structure from sources like the Protein Data Bank (PDB). For our specific case, we are docking sinigrin in myrosinase from Sinapis alba. This involves the myrosinase protein with bound gluco-hydroximolactam and sulfate (PDB ID: 1E6S). Please note, the example provided focuses on docking sinigrin in myrosinase from Sinapis alba with bound gluco-hydroximolactam and sulfate. Additionally, these procedures are outlined to be used in a Windows or a Linux machine. 2.1 Create a Working Directory:Establish a directory on your local drive. For instance: F:\Dropbox\Methods\docking. Example command (for making a directory in Linux) mkdir Dropbox\Methods\docking 2.2 Download and Rename Receptor File:Download the receptor file (1E6S.pdb) into your working directory and rename it to receptor.pdb. Example command (for downloading and renaming in Linux): wget https://www.rcsb.org/structure/1e6s mv 1E6S.pdb receptor.pdb 2.3 Clean the Receptor File:2.3.1 Remove specified lines from the receptor.pdb file:Keep lines containing ATOM, HETATM, and TER. On Linux, execute the following command. In Windows, you have to do it manually. cat receptor.pdb | egrep "^ATOM|^HETATM|^TER" > t.pdb \mv t.pdb receptor.pdb 2.3.2 Clean Crystal Waters and Hetero Groups:Remove crystal water (HOH) lines and all hetero groups from the end of the receptor.pdb file. Post-docking, you can relocate crystal waters, eliminating those with short contacts either with each other or other residues. This step is advisable, especially if planning QM/MM calculations. Note: Retention of metal ions situated in the active site might be necessary for certain proteins. This process ensures the receptor.pdb file is streamlined for docking, allowing flexibility to adjust crystal waters and hetero groups after the initial docking phase. cat receptor.pdb | egrep "^ATOM" > t.pdb \mv t.pdb receptor.pdb 2.4 Isolate the Test-Ligand Information:Extract lines with the identifier "GOX" from the 1E6s.pdb file and copy them into a new text file named test-ligand.pdb. The GOX group, mimicking sinigrin binding in the receptor's active site [5], is essential for defining the search space in section 3.1.4. For Linux, execute the following command: grep 'GOX' 1E6s.pdb > test-ligand.pdb The test-ligand.pdb file should contain the GOX group details, while the receptor.pdb is cleaned and ready for subsequent steps in the docking procedure. You will get the following in the test-ligand.pdb file. HET GOX M 921 13 HETNAM GOX (2S,3S,4R,5R)-6-(HYDROXYAMINO)-2-(HYDROXYMETHYL)-2,3,4, HETNAM 2 GOX 5-TETRAHYDROPYRIDINE-3,4,5-TRIOL HETSYN GOX D-GLUCONHYDROXIMO-1,5-LACTAM FORMUL 11 GOX C6 H12 N2 O5 HETATM 4334 C1 GOX M 921 46.871 119.891 52.844 1.00 12.77 C HETATM 4335 N1 GOX M 921 46.180 119.101 52.013 1.00 14.84 N HETATM 4336 C2 GOX M 921 47.752 119.220 53.910 1.00 11.14 C HETATM 4337 N5 GOX M 921 46.714 121.224 52.640 1.00 13.32 N HETATM 4338 O7 GOX M 921 45.512 119.568 50.983 1.00 19.98 O HETATM 4339 O2 GOX M 921 47.199 118.054 54.530 1.00 11.49 O HETATM 4340 C3 GOX M 921 48.249 120.259 54.889 1.00 11.04 C HETATM 4341 O3 GOX M 921 49.347 119.649 55.564 1.00 11.04 O HETATM 4342 C4 GOX M 921 48.648 121.507 54.062 1.00 12.38 C HETATM 4343 O4 GOX M 921 49.290 122.463 54.907 1.00 11.97 O HETATM 4344 C5 GOX M 921 47.312 122.173 53.572 1.00 13.58 C HETATM 4345 C6 GOX M 921 47.406 123.456 52.773 1.00 13.67 C HETATM 4346 O6 GOX M 921 48.187 123.278 51.578 1.00 15.18 O
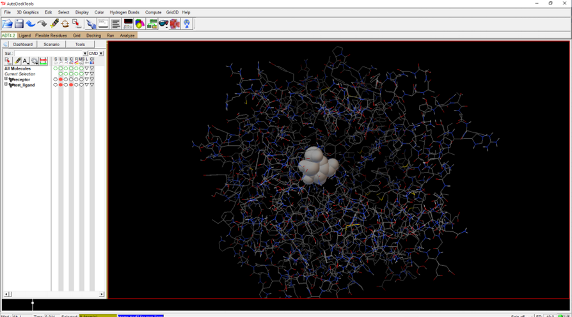
3. Converting Files to Vina Format and Setting Up Docking Calculations3.1 Preparing the Receptor:3.1.1 Launch MGLTools and open the receptor.pdb file (File/Read Molecule) from your working directory.3.1.2 Add polar hydrogens to the receptor (Edit/Hydrogens/Add/Polar Only). Docking requires the addition of polar hydrogens.3.1.3 Save the modified receptor.pdb file in pdbqt format (Grid/Macromolecule/Choose/Protein/Select). Save this as receptor.pdbqt in your working directory.3.1.4 Setting Up the Search Space:3.1.4.1 Open the ligand-test.pdb file in MGLTools (Ligand/Input/Open). Figure 1 shows the test ligand inside the receptor.
Figure 1. The test ligand (space fill in the center), inside the receptor.
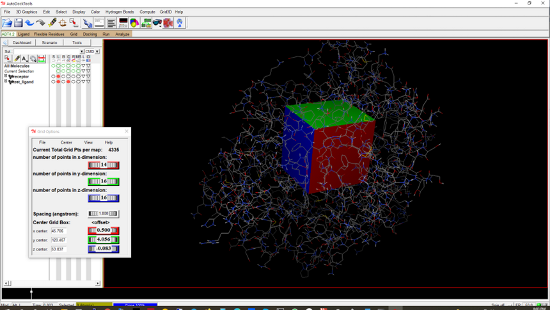
3.1.4.2 Click Grid/Grid Box to open Grid Options (refer to Figure 2).3.1.4.3 Set measurements to angströms. To do this, hold down the left mouse button on the fourth rectangular box (the black rectangle in Figure 2) and move the mouse to the right-hand side until the value is 1.0.3.1.4.4 Adjust the box to enclose the test ligand. MGLTools displays the search space with red, green, and blue boxes in the x, y, and z directions, respectively. Adjust by dragging the mouse in the Grid Options window.3.1.4.5 Note down the x, y, and z sizes, center x, center y, and center z values for later use in AutoDock Vina (4.3). Figure 2. The receptor and the search space around the test ligand. The Grid Options window is shown on the left-hand side.
3.1.5 Converting the Ligand (Substrate) structure to the pdbqt Format:
If the ligand lacks hydrogens, add polar-only hydrogens (Edit/Hydrogens/Add/Polar Only) and compute Gasteiger charges (Edit/Charges/Compute Gasteiger). Here's an example of the optimized PDB structure of sinigrin. HETATM 1 O4 CGT 503 -2.756 -7.478 7.839 O HETATM 2 H11 CGT 503 -2.899 -6.536 7.635 H HETATM 3 C6 CGT 503 -3.195 -8.124 6.636 C HETATM 4 H9 CGT 503 -2.782 -9.134 6.591 H HETATM 5 H10 CGT 503 -4.284 -8.211 6.672 H HETATM 6 C5 CGT 503 -2.791 -7.320 5.383 C HETATM 7 O9 CGT 503 -2.755 -5.900 5.749 O HETATM 8 H8 CGT 503 -3.549 -7.545 4.628 H HETATM 9 C4 CGT 503 -1.413 -7.685 4.784 C HETATM 10 O3 CGT 503 -1.253 -9.067 4.471 O HETATM 11 H7 CGT 503 -0.860 -9.055 3.580 H HETATM 12 H6 CGT 503 -0.619 -7.381 5.470 H HETATM 13 C3 CGT 503 -1.314 -6.892 3.470 C HETATM 14 O2 CGT 503 -0.138 -7.225 2.740 O HETATM 15 H5 CGT 503 0.214 -6.376 2.418 H HETATM 16 H4 CGT 503 -2.190 -7.115 2.855 H HETATM 17 C2 CGT 503 -1.247 -5.402 3.815 C HETATM 18 O1 CGT 503 -1.190 -4.775 2.536 O HETATM 19 H3 CGT 503 -1.897 -5.226 2.040 H HETATM 20 H2 CGT 503 -0.314 -5.210 4.351 H HETATM 21 C1 CGT 503 -2.429 -4.920 4.723 C HETATM 22 H1 CGT 503 -3.339 -4.714 4.139 H HETATM 23 S1 CGT 503 -1.928 -3.396 5.623 S HETATM 24 C10 CGT 503 -2.333 -3.542 7.323 C HETATM 25 C7 CGT 503 -3.685 -3.028 7.764 C HETATM 26 C8 CGT 503 -3.757 -1.526 7.910 C HETATM 27 C9 CGT 503 -4.834 -0.802 7.653 C HETATM 28 H15 CGT 503 -4.819 0.278 7.780 H HETATM 29 H16 CGT 503 -5.793 -1.238 7.386 H HETATM 30 H14 CGT 503 -2.835 -1.040 8.219 H HETATM 31 H12 CGT 503 -3.992 -3.519 8.690 H HETATM 32 H13 CGT 503 -4.340 -3.355 6.953 H HETATM 33 N1 CGT 503 -1.542 -3.977 8.252 N HETATM 34 O5 CGT 503 -0.263 -4.418 7.792 O HETATM 35 S2 CGT 503 1.039 -3.633 8.294 S HETATM 36 O7 CGT 503 0.593 -2.273 8.465 O HETATM 37 O8 CGT 503 1.455 -4.267 9.516 O HETATM 38 O6 CGT 503 2.008 -3.778 7.252 O END
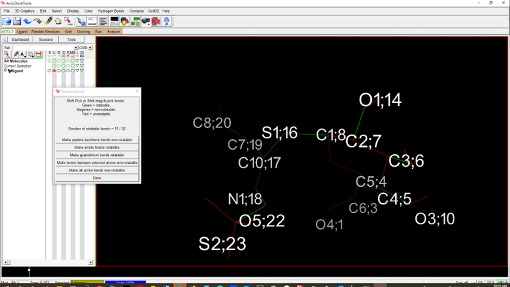
3.1.5.1 Restart MGLTools.3.1.5.2 Open the ligand.pdb file (Ligand/Input/Open) in MGLTools to add Gasteiger charges and detect rotatable bonds automatically.3.1.5.3 Optionally edit rotatable bonds (Ligand/Torsion Tree/Choose Torsions). Green-colored bonds are rotatable, while pink and green represent nonrotatable ones (refer to Figure 3).3.1.5.4 Save the ligand structure as ligand.pdbqt (Ligand/Output/Save as PDBQT). Exit MGLTools after saving.
Figure 3. Representation of the ligand in MGLTools. The rotatable and nonrotatable bonds are shown in green and red, respectively. 4. Docking by AutoDock Vina4.1 Windows users can open the command prompt by searching for "cmd" in the toolbar, while Linux users can open a terminal.4.2 Navigate to the working directory (e.g., cd \Dropbox\Methods\docking). Switch to another drive by typing the drive letter followed by ":" (e.g., "F:") to access that drive.4.3 Create a configuration file (conf.txt) in the working directory with the docking options. Use the x, y, and z sizes, center x, center y, and center z values obtained from step 3.1.4.5. Here's an example conf.txt file:receptor = receptor.pdbqt ligand = ligand.pdbqt out = out.pdbqt center_x = 43.223 center_y = 118.899 center_z = 54.290 size_x = 16 size_y = 14 size_z = 10 cpu = 4 num_modes = 10 energy_range = 3
4.4 Run the docking calculations by calling AutoDock Vina:On Windows: Open Command Prompt and navigate to the AutoDock Vina installation directory. Then run the command: "C:\vina\vina.exe" --config conf.txt --log log.txt On Linux: Run the command: vina --config conf.txt --log log.txt 5. Analyzing Output Files:5.1 Convert the out.pdbqt file to out.pdb using OpenBabel. In Linux, use the command:babel -i pdbqt out.pdbqt -o pdb out.pdb You can split the binding modes using the vina_split program before the conversion: On windows: "C:\vina\vina_split.exe" --input out.pdbqt On Linux: vina_split --input out.pdbqt 5.2 Visualize the binding modes inside the receptor:The out.pdb file includes structures of binding poses of the docked ligand inside the receptor. To visualize specific binding modes, copy the PDB structure from out.pdb into the receptor.pdb file and open it in visualization software like Rasmol. MGLTools can also help in visualizing binding modes (Analyze/Docking/Open Auto Dock Vina Results/out.pdbqt/Multiple molecules, and then Grid/Macromolecule/Open/receptor.pdbqt). Refer to Figure 4 for an example of one binding mode inside the enzyme.
Figure 4. One of the binding modes inside the enzyme. 6. Exercise:Locate the Active Sites and Perform Docking of Xevinapant (PubChem CID: 25022340) into the active sites of specified receptors. Receptors and PDB IDs: LC3-p62 complex (PDB ID: 2ZJD) Beclin 1 evolutionarily conserved domain (PDB ID: 4DDP) CIAP1-BIR3 in complex with N-terminal peptide from Caspase-9 (PDB ID: 3D9T) Receptor-interacting serine/threonine-protein kinase 3 (AlphaFold: Q9Y572) 7. References[1] O. Trott, A.J. Olson, Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem. 31 (2010) 455–461. https://doi.org/10.1002/jcc.21334. [2] G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell, A.J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem. 30 (2009) 2785–2791. https://doi.org/10.1002/jcc.21256. [3] N.M. O’Boyle, M. Banck, C.A. James, C. Morley, T. Vandermeersch, G.R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminform. 3 (2011) 33. https://doi.org/10.1186/1758-2946-3-33. [4] M.D. Hanwell, D.E. Curtis, D.C. Lonie, T. Vandermeersch, E. Zurek, G.R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminformatics 2012 41. 4 (2012) 1–17. https://doi.org/10.1186/1758-2946-4-17. [5] W.P. Burmeister, S. Cottaz, P. Rollin, A. Vasella, B. Henrissat, High resolution X-ray crystallography shows that ascorbate is a cofactor for myrosinase and substitutes for the function of the catalytic base., J. Biol. Chem. 275 (2000) 39385–93. https://doi.org/10.1074/jbc.M006796200.
|



|
Department of Chemistry, Faculty of Science, University of Kurdistan |

|
Mehdi Irani Teaching duties Methods |